This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison.
What is a phylogeny?
A phylogeny, or phylogenetic tree, is a diagram that shows lines of evolutionary decent of species, in our case protein sequences of species, from a common ancestor. These trees are useful to see the evolutionary relationship of species and to see how different species may have split during evolution based on gene flow, speciation, and isolation, from a common ancestor [1]. There are many different ways to generate trees, often resulting in different branching. Each tree represents a hypothesis of how the species could have diverged from a common ancestor, therefore many different trees and hypothesis can exist. Getting the same or very similar trees using different methods of generation, suggests the tree is more accurate [1].
Understanding phylogenetic trees
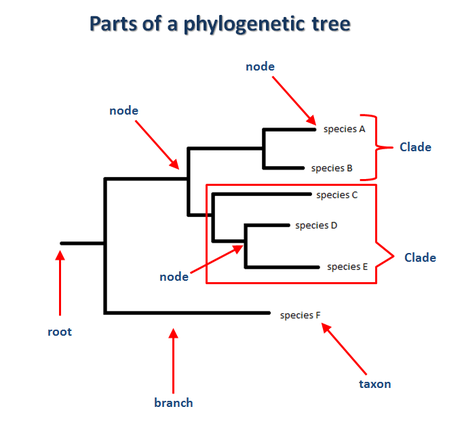
Root: The common ancestor of all descendant species in the phylogeny
Taxon: A group of organisms, often a species, that has descended from a common ancestor
Branch: Represents a divergence from a common ancestor into new species; each branch represents a new lineage
Node: A node represents a common ancestor of the species that follow it
Clade: Two or more taxa that includes the common ancestor and all of the descendants
Outgroup: A taxon that is less related to the group of interest; it branches off at the base of the tree. Species F is an example in this diagram [16].
Taxon: A group of organisms, often a species, that has descended from a common ancestor
Branch: Represents a divergence from a common ancestor into new species; each branch represents a new lineage
Node: A node represents a common ancestor of the species that follow it
Clade: Two or more taxa that includes the common ancestor and all of the descendants
Outgroup: A taxon that is less related to the group of interest; it branches off at the base of the tree. Species F is an example in this diagram [16].
For more information about phylogenetic trees, please consider viewing the following video
BTBD9 protein phylogeny
The phylogenetic trees below were created using the homologous protein sequences of the human BTBD9 protein of 12 different species, including homo sapiens. In the phylogenetic trees shown below, the first two were made using ClustalW2 which is a program that uses a method called neighbor joining to create trees. Neighbor joining is a method that constructs phylogenetic trees from evolutionary distance data. The goal is to minimize branch length based on the minimum evolution between the species. Computer models have found this method to be quite efficient at producing the correct tree layout, (recall, however, that trees are hypothesis, so do not necessarily state for certain the exact evolutionary path) but the algorithm it is based off of is quite complex and more than is necessary for our purposes [2]. The Neighbor joining method puts together groups with the fewest amino acid differences in their protein sequence [15]. From left to right in the images below: the first tree was made using the neighbor joining method, based on percent identity of the protein sequence, the second tree was made using the neighbor joining method and BLOSUM62. Blosum, Blocks Substitution Matrix, is a method to generate phylogeny trees that is based on observations of the frequencies of substitutions in local alignments of related proteins. BLOSUM62 creates a phylogeny in which there is no more than 62% identity between protein sequences. Sequences over 62% identical are considered one sequence and represented as such in the comparison [3]. The third tree was made using the program T-Coffee which also uses the neighbor joining method[4]. If you would like to see or use the annotated FASTA sequences I used to generate these trees, please download the following file. Please click on the trees to make them larger
| annotatedfastas.pdf |
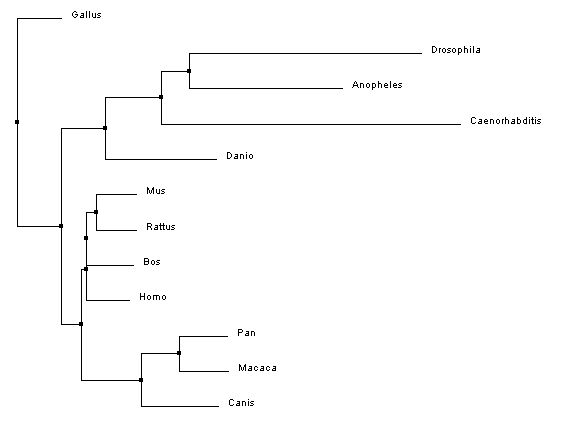
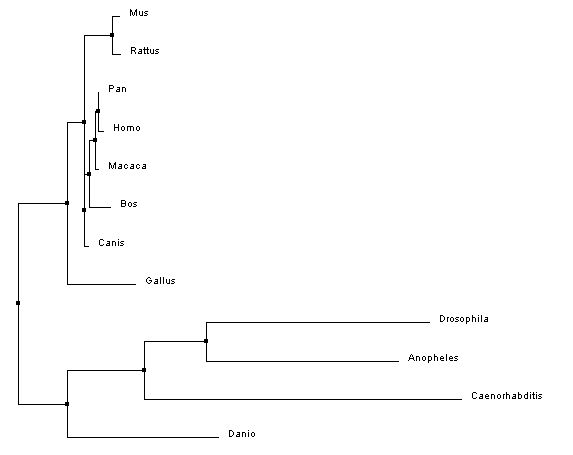
Analysis and discussion
In the three phylogenetic trees above, different trees arise from the use of different programs and conditions to generate the trees. The trees are not consistent due to varying factors such as horizontal gene transfer and differing rates of evolution [7]. All three trees are really quite different, with a similarity that they all have a clade made up of Drosophila melanogaster, Anopheles gambiae, Caenorhabditis elegans, and Danio rerio in which they all show the same relationship in relatedness. This clade also had the lowest percent identities with the homologous BTBD9 human protein sequence, shown on the protein Homology page.
Trees with the highest percent maximum identity
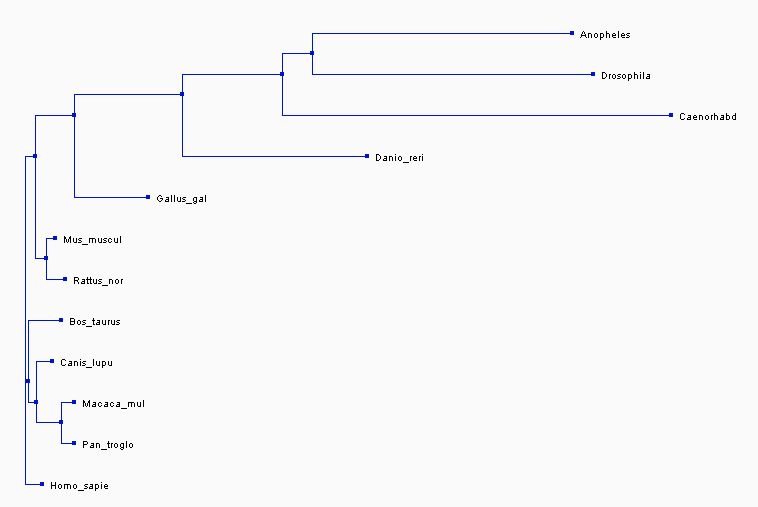
The first two trees shown below were created in the exact same way as the first two trees shown above, except that they show three fewer species and thus are different trees. The third tree is constructed using the maximum likelihood method on Phylogeny.fr [8-13]. The Maximum likelihood method is used to construct the most likely phylogenetic tree given a specific model for molecular evolution and a data set [14].
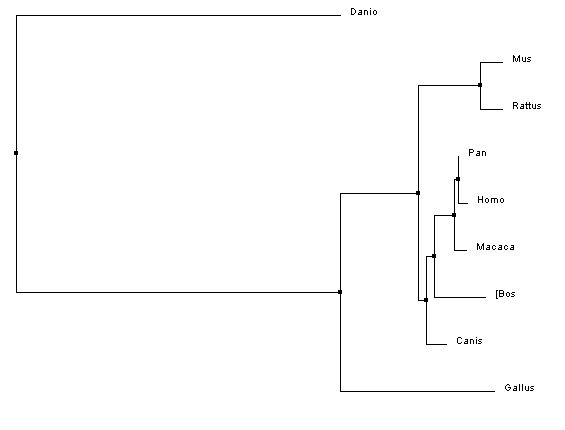
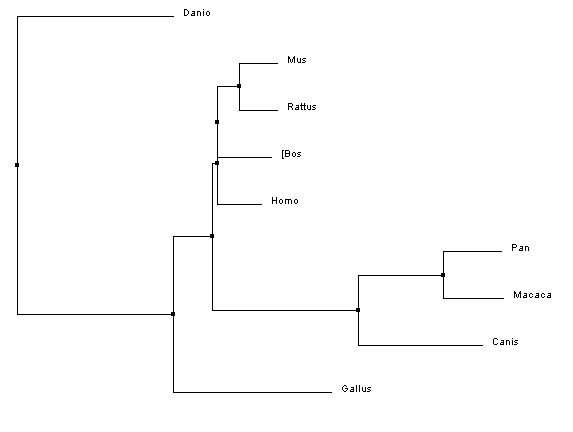
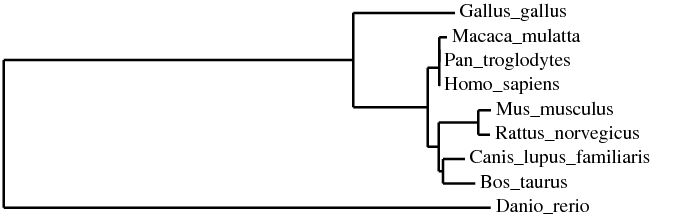
Analysis and discussion
In all three of the above phylogenetic trees, Danio rerio is an out group as is Gallus gallus, although not quite as separated. In the neighbor joining method with percent identity and the phylogeny.fr tree, Homo sapies, Pan troglodytes, and Macaca mulatta are grouped into a clade. In the two different neighbor joining trees Mus musculus and Rattus norvegius are grouped into a clade.
References
[1] Baum, D. (2008) Reading a phylogenetic tree: The meaning of monophyletic groups. Nature Education 1(1)
[2] Saitou, N., Nei, M. (1987). The neighbor-joining method: A new method for re-constructing phylogenetic trees. Molecular biology and evolution, 4(4): 406-25. Retrieved March 6, 2013, from http://www.ncbi.nlm.nih.gov/pubmed/3447015.
[3] http://www.ncbi.nlm.nih.gov/books/NBK62051/
[4] http://www.ebi.ac.uk/Tools/msa/tcoffee/
[5] ClustalW and ClustalX version 2 (2007). Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ and Higgins DG. Bioinformatics 2007 23(21): 2947-2948. doi:10.1093/bioinformatics/btm404
[6] A new bioinformatics analysis tools framework at EMBL-EBI (2010). Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. Nucleic acids research 2010 Jul, 38 Suppl: W695-9. doi:10.1093/nar/gkq313
[7] Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999 Jan;21(1):108-10. PMID: 9916801
[8] Dereeper A., Audic S., Claverie J.M., Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010 Jan 12;10:8. (PubMed)
[9] Dereeper A.*, Guignon V.*, Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W465-9. Epub 2008 Apr 19. (PubMed) *: joint first authors
[10] Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, Mar 19;32(5):1792-7. (PubMed)
[11] Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, Apr;17(4):540-52. (PubMed)
[12] Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, Oct;52(5):696-704. (PubMed)
[13] Anisimova M., Gascuel O. Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Syst Biol. 2006, Aug;55(4):539-52. (PubMed)
[14] Zimmer, C., & Emlen, D. J. (2013). Evolution making sense of life. (pp. 262-263). Greenwood Village, CO: Roberts and Company Publishers, Inc.
[15] http://vimeo.com/829413
[16] http://evolution.berkeley.edu/evolibrary/article/phylogenetics_02
Header photo: https://commons.wikimedia.org/wiki/File:Phylogenetic_Tree_of_Life.png
[2] Saitou, N., Nei, M. (1987). The neighbor-joining method: A new method for re-constructing phylogenetic trees. Molecular biology and evolution, 4(4): 406-25. Retrieved March 6, 2013, from http://www.ncbi.nlm.nih.gov/pubmed/3447015.
[3] http://www.ncbi.nlm.nih.gov/books/NBK62051/
[4] http://www.ebi.ac.uk/Tools/msa/tcoffee/
[5] ClustalW and ClustalX version 2 (2007). Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ and Higgins DG. Bioinformatics 2007 23(21): 2947-2948. doi:10.1093/bioinformatics/btm404
[6] A new bioinformatics analysis tools framework at EMBL-EBI (2010). Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. Nucleic acids research 2010 Jul, 38 Suppl: W695-9. doi:10.1093/nar/gkq313
[7] Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999 Jan;21(1):108-10. PMID: 9916801
[8] Dereeper A., Audic S., Claverie J.M., Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010 Jan 12;10:8. (PubMed)
[9] Dereeper A.*, Guignon V.*, Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W465-9. Epub 2008 Apr 19. (PubMed) *: joint first authors
[10] Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, Mar 19;32(5):1792-7. (PubMed)
[11] Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, Apr;17(4):540-52. (PubMed)
[12] Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, Oct;52(5):696-704. (PubMed)
[13] Anisimova M., Gascuel O. Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Syst Biol. 2006, Aug;55(4):539-52. (PubMed)
[14] Zimmer, C., & Emlen, D. J. (2013). Evolution making sense of life. (pp. 262-263). Greenwood Village, CO: Roberts and Company Publishers, Inc.
[15] http://vimeo.com/829413
[16] http://evolution.berkeley.edu/evolibrary/article/phylogenetics_02
Header photo: https://commons.wikimedia.org/wiki/File:Phylogenetic_Tree_of_Life.png

{kind=link}